This article was published in Scientific American’s former blog network and reflects the views of the author, not necessarily those of Scientific American
Microbes are all around us, not to mention all up in us, but finding and characterizing them is not a trivial task. But it’s gettingeasier. As I wrote about last year, advances in DNA sequencing technology mean that we are no longer restricted to isolating individual microbes, growing them up in a lab and studying them one by one. We can instead isolate the DNA from a particular environment, and just sequence everything that’s there. But a paper in the new journal Nature Microbiology suggests the most common method for analyzing new microbial populations is systematically missing a substantial fraction of what’s there.
Metagenomics uncovers gaps in amplicon-based detection of microbial diversity
Unless you’re a microbiologist, you may need a primer on two terms from the title - “metagenomics,” and “amplicon-based detection.” I give them a more complete treatment in that series from last year, but I’ll recap briefly. A useful metaphor for this sort of sequencing approach is walking into a library and trying to catalogue its contents. Amplicon sequencing is like scanning the spines of the books and just recording their titles, while metagenomic sequencing is like going through and recording all of the text inside all of the books. Of course, you can imagine which of these approaches is easier.
On supporting science journalism
If you're enjoying this article, consider supporting our award-winning journalism by subscribing. By purchasing a subscription you are helping to ensure the future of impactful stories about the discoveries and ideas shaping our world today.
The metaphor isn’t perfect, since genomes don’t have “titles” readily accessible. Instead, scientists know the general form that titles take, and can amplify those sequencing using a technique called the polymerase chain reaction or PCR. What this paper shows is that the so-called “universal” primers that scientists have been using aren’t as universal as we thought. It’s like we’ve only been looking for titles that start with letters, and in many cases completely missed Orwell’s 1984.
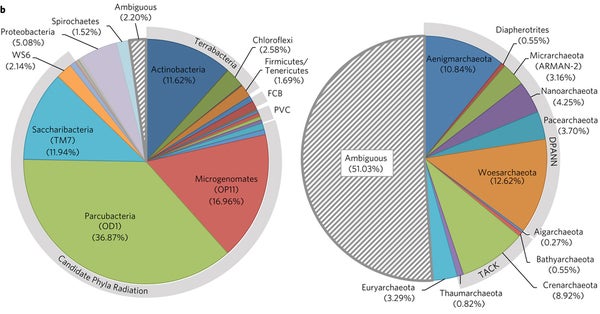
Emiley A. Eloe-Fadrosh and colleagues analyzed over 6000 metagenomic data sets, and checked to see how many of the SSU genes (the ones used as titles for the genome) would have been detected by the commonly used microbial primers. They found that 10% or more would have been missed entirely. And not surprisingly, most of those bacteria that would be missed are members of novel or at least very under-studied species.

Figure 1a - % of microbes in a metagenomic sample that would be missed by the given primers.
Unfortunately, the fix for this is not readily apparent. The authors point to single-cell and deep metagenomic sequencing, but these techniques are still comparatively expensive, especially when it comes to the expertise and computational infrastructure required to analyze the reams of data returned. There’s also a huge gap in functionally characterizing these hoards of new bugs - just because we know they’re there, it doesn’t mean we know what they do. Still, knowing they’re there is a necessary first step.